Evolving Our Assessment & Future Guiding Principles Workshop Report (2023)
On this page
Skip the menu of subheadings on this page.
Workshop report main page logo. An dark outlined circle in light blue with the COT logo at the top with dark text underneath.
Background and Objectives
1. The Committee on Toxicity of Chemicals in Food, Consumer Products and the Environment (COT) held a workshop to start work on updating their guidance on toxicity testing and its supporting principles. The starting point for the process is to use existing frameworks and guidance but with the aim of introducing innovative improvements where appropriate.
2. The workshop aimed to identify areas where guidance needs to evolve and included reviewing fundamental risk assessment principles, current guidance on risk assessment and what can be learned from it, integration of new approach methodologies, exploring hazard vs risk and weight of evidence.
3. The overall objective was to discuss how the Committee moves forward in a new era of risk assessment.
Workshop Overview
4. The workshop took place in May 2023 in London, United Kingdom. The workshop included themed sessions consisting of short flash presentations followed by roundtable discussion. The four sessions were: Where are we at; What we need to improve; How to achieve; and Looking to the future - moving forward.
Introductions and aims of the day: Objectives and Overall Principles
5. Professor Boobis introduced the workshop, set the scene by outlining the objectives and gave an introduction to the COT, its guidelines and principles.
6. The COT was established in 1978. The COT is a committee of independent experts that provides advice to the Food Standards Agency (FSA), the Department of Health and Social Care (DHSC) and other Government Departments and Agencies on matters concerning the toxicity of chemicals in food, consumer products and the environment. Even though the Committee largely advises on chemicals in food, the COT has a wide remit and advises various government departments on matters including consumer products such as e-cigarettes. The remit is so broad that in future, the committee may want to revisit and clarify it, as some of the topics are covered under the remits of other scientific advisory committees. The Committee covers such a wide range of chemicals that, in principle, essentially any chemical that humans are exposed to comes under its terms of reference.
7. The methodologies and approaches needed for risk assessment should be broad and flexible to cover the variety of topics and questions the COT may be required to address.
8. The current COT guidance, which was called “guidelines” at the time of preparation, was published in 1982, and has never been updated since. There is a lot of text regarding animal husbandry and different test types. This has now been superseded by standardised test guidelines that COT adopt or fully endorse, which are utilised elsewhere.
9. The focus for the workshop should be on the principles. The experts that put together the 1982 general guidelines were quite foresighted stating for example: “It would be undesirable to draw up an inflexible list of tests which need to be carried out on every chemical” as well as the importance of exposure assessment: “Consideration of the safety-in-use of a particular chemical should be based on the likely routes, levels and duration of exposure in man and on comparative pharmacokinetic and metabolic data, if available”. Interestingly, the rationale for the use of animals is mentioned: “The results of studies in animals normally form the major part of the evaluation... Although this is not an ideal situation, the use of animals for toxicity testing provides the best means currently available” highlighting that it was using the best available science at that time. Mode of action was also introduced “It is now recognised that a variety of non-specific factors may influence the incidence of neoplasms in aged animals…evidence to support this possibility should be sought” highlighting that the guidelines have stood the test of time. Other chapters were mentioned such as toxicological data requirements.
10. Since the guidelines were published, there has been a lot of work in the area of risk assessment and a lot of developments, not just in technology but also in the way evidence synthesis, modelling, integration of evidence, and systematic reviews are approached.
11. The speaker then emphasised there was no need to reinvent the wheel and existing frameworks like the World Health Organisation (WHO) Principles and methods for the risk assessment of chemicals in food (2009) and the European Food Safety Authority (EFSA) Guidance and other assessment methodology documents should be used or adapted as necessary.
12. The risk analysis process from the general principles document was then discussed, which includes risk assessment, risk management and risk communication. Problem formulation will be the key interface between the scientific advisory committee (SAC) and risk management i.e., is the right question being asked to be able for the SAC to give meaningful advice.
13. Professor Boobis then gave personal thoughts and recommendations on some areas of the guidance that might require consideration, which included identifying points of departure (POD), including modelling aspects, derivation of health-based guidance values (HBGVs), uncertainty factors and new approach methodologies (NAMs) when not using traditional animal data. The whole area of mode of action and adverse outcome pathways (AOPs) has evolved, with assessment of the key biological steps through to an adverse outcome using a variety of methods, including in silico approaches to micro-physiological systems, and this needs to be taken into consideration. The COT / FSA ongoing roadmap on NAMs has highlighted these areas and how to extrapolate or determine a HBGV, the relationship between adversity and non-adverse outcomes as well as exposure duration profiles should be considered. Finally, it will be important to consider how the guidance will address exposure assessment and the influence of the microbiome as well as evidence integration.
14. Professor Boobis concluded that this is the start of a process of data interpretation and that the guidance and general principles should be pragmatic where possible to ensure their longevity.
Session 1: Where are we at? The good, the bad and the ugly
What do we currently have (what is the EFSA process/guidelines), how it is evolving?
Chemical Mixtures
15. Professor Christer Hogstrand presented on the assessment of chemical mixtures. The speaker started off by stating the areas of relevance for EFSA:
- Human Risk Assessment which includes regulated products, food additives, feed additives, food contact materials, pesticides and contaminants in food and feed chain.
- Animal Risk Assessment which includes pesticides, feed additives and contaminants in feed.
- Ecological Risk Assessment which includes pesticides, feed additives and contaminants in food and feed chain.
16. The speaker discussed harmonisation of the different methodologies and processes in different areas, noting these were similar and could be applied to chemical mixtures across the different areas of risk assessment process.
17. EFSA have created some guidance documents: Guidance on harmonised methodologies for human health, animal health and ecological risk assessment of combined exposure to multiple chemicals and Guidance Document on Scientific criteria for grouping chemicals into assessment groups for human risk assessment of combined exposure to multiple chemicals.
18. It was discussed that for combined chemical risk assessment, grouping is the hardest part.
19. The way EFSA was dealing with interactions (synergism or antagonism) was to apply an additional uncertainty factor only if there were indications of possible interactions at relevant exposure levels.
20. The size of the factor used should be determined on a case-by-case basis depending on the strength of the evidence for the presence or absence of interactions, the expected impact of the interactions and the level of conservativeness in the assessment.
21. Most of the risk assessment was based on an overarching framework for chemical risk assessment consisting of problem formulation, exposure assessment, hazard assessment and risk characterisation.
22. There were a couple of ways to conduct mixtures risk assessment: Whole mixtures approach; consider the mixture as if it was a single chemical. This is thought to be a sensible way of doing things, the idea being that people are exposed to the mixture so essentially exposed to an individual entity, but it is complicated. There are several problems and challenges with this approach. Examples include: defining what the chemical mixture is; how do you know that the chemical mixture sold currently as a product is the same as the one generated 5 years ago? are there different ratios between the different components? At a low concentration one chemical substance could have effects by itself but you could miss the effects if tested as a whole mixture. Therefore, the most scientific approach was a component-based approach. You assess each of the different components and then bring the information on them together and add them up in the risk characterisation together with exposure assessment.
23. For each of the steps in the risk assessment you apply a step wise approach.
24. The stepwise approach for hazard identification and characterisation using a component-based assessment is guided by dos/ don’ts to be considered as part of the process (Cattaneo et al., 2023).
25. Step 1: risk assessors have the opportunity to confirm or refine the initial grouping of chemicals performed at the problem formulation stage. If needed, a refinement can be made using WoE approaches, dosimetry (toxicokinetics) or mechanistic data (i.e., Mode of Action (MoA and AOP).
26. Step 2: the relevant entry tier for the assessment is decided based on the purpose of the assessment and the available data. Hazard information is collected for each individual chemical and may include toxicity data, reference points, reference values, mechanistic data, toxicokinetic information and relative potency information. In case of data-poor situations, a list of possibilities to fill data gaps is identified.
27. Step 3: assesses the evidence for independent action between individual chemicals of the assessment groups and the potential for interactions. Within step 3, the most appropriate approach for risk characterisation is defined.
28. Step 4: for each individual component of the assessment group, reference point and uncertainty factors are derived to obtain appropriate reference values through the relevant tier. Such reference values can be used for individual components of the whole group (equivalents of an index chemical).
29. Step 5: summarises the hazard metrics for individual components and lists assumptions and uncertainties.
30. The speaker then discussed the Guidance Document on Scientific criteria for grouping chemicals into assessment groups for human risk assessment of combined exposure to multiple chemicals.
31. Whenever available, AOP information should be used to define assessment groups and for grouping chemicals (OECD, 2018).
32. There can be different Molecular Initiating Events (MIE) that need to converge into key events then end up as a specific AOP. Chemicals that share a common adverse outcome and for which their AOPs are known, should be grouped together in the same assessment group. This approach, which embraces a range of chemicals covering one or more different AOPs, may involve chemicals that trigger: the same AOP by interacting with the same MIE; separate AOPs which then converge at any intermediate key event; an AOP which leads to the same adverse outcome without converging at intermediate key event from other AOPs.
33. The EFSA Committee noted that these three categories include all chemicals with the same adverse outcome but distinct MIEs, thus having comprehensive mechanistic understanding. Such mechanistic information anchored to a Mode of Action (MoA), AOP or its related network allows the uncertainty of the chemical grouping to be reduced. However, if the available evidence indicates that chemicals with a common MoA do not contribute to the combined effects based on exposure and potency considerations, these may be excluded from the final assessment group.
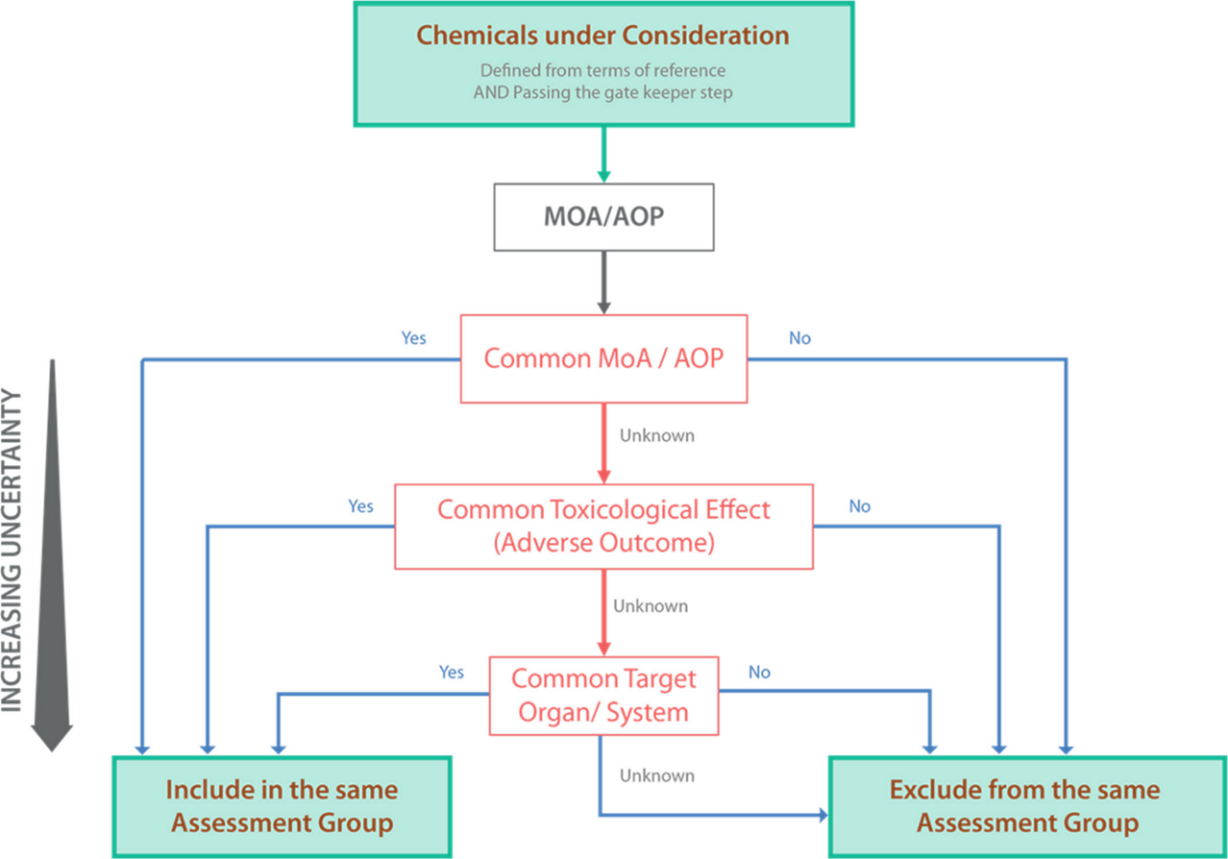
A multi coloured diagram showing a flow chart top-down hierarchical process for grouping chemicals into assessment groups using hazard-driven criteria. The thickest arrow indicates the gold standard hazard-driven criteria (MoA/AOP) with the lowest uncertainty. The flow chart comprises of solid colour boxes and outlines with different coloured text. The directional arrows are in blue, green, red and black.
Figure 1. Top-down hierarchical process for grouping chemicals into assessment groups using hazard-driven criteria. The thickest arrow indicates the gold standard hazard-driven criteria (MoA/AOP) with the lowest uncertainty.
34. The scheme (Figure 1) that was created is a hierarchical framework to apply hazard-driven criteria for the grouping of chemicals into assessment groups using available mechanistic information (MoA or AOP) as the gold standard. The top-down hierarchical framework supports generating such mechanistic data and reducing uncertainty in the grouping process.
35. One of the challenges of this approach is that one receptor leads to several critical effects. MIEs that have divergent outcomes, e.g. activation of the Aryl Hydrocarbon Receptor (ARH) by dioxin and compounds with dioxin-like toxicity can then lead to a whole range of outcomes and for some of them we know the AOP and for some of them we know little to nothing. Nevertheless, as they share the same MIE, the adverse effects are related, and it can be argued that they should be grouped even if we don’t have data on the same adverse outcome for the different members of this group. Therefore, it would be a case-by-case basis decision.
36. The speaker then described another issue using the example of Perfluoroalkyl and Polyfluoroalkyl Substances (PFAS). EFSA assessed the effects of the chemicals based on epidemiological studies showing apical effects and no knowledge of AOP, only that there is an effect for a group of chemicals. This was the case for the EFSA Risk to human health related to the presence of perfluoroalkyl substances in food perfluoroalkyl substances in food. The general conclusions were presented:
- “It was considered that similarities in chemical properties and effects warrant a mixture approach for a number of PFASs…. Also, in terms of effects, these compounds in general show the same effects when studied in animals.”
- “Based on several similar effects in animals, toxicokinetics and observed concentrations in human blood, the Panel on Contaminants in the Food Chain (CONTAM) decided to perform the assessment for the sum of four PFASs: Perfluorooctanoic acid (PFOA), Perfluorononanoic acid (PFNA), Perfluorohexanesulphonic acid (PFHxS) and Perfluorooctane sulfonate (PFOS)…Equal potencies were assumed for the four PFASs included in the assessment.”
- “Based on available studies in animals and humans, effects on the immune system were considered the most critical for the risk assessment…. Since accumulation over time is important, a tolerable weekly intake (TWI) of 4.4 ng/kg bw per week was established.”
37. The problem in the speaker's personal view was that the studies were done in the where individuals were exposed not only to PFAS but to a whole range of chemicals from the available food. Although this was a problem with the Faroe Islands study, there was another study from Germany, which supported the same effects (Kotthoff et al., 2020). Effects on the immune system were the critical effects and this was probably because the molecules resemble lipids, acting as inflammatory agents, but the AOP or MoA associated with the critical effect on antibody titre against different vaccines was unknown. Again, the speaker emphasised that this was his personal view.
38. Moving forward, EFSA were aiming to develop and implement a New Approach Methodologies (NAMs)-based Integrated Approaches to Testing and Assessment (IATA) for exploring the mode of action for the observed immunosuppressing effects and for addressing immunotoxicity of PFAS, such as PFOS and PFOA, including the assessment of a common MoA and differences in potencies.
Weight of Evidence
39. Professor Maged Younes presented on Weight of Evidence (WoE).
40. EFSA Guidance on the use of the weight of evidence approach in scientific assessments was developed by EFSA, together with guidance documents on Biological Relevance and on Uncertainty, with the aim of increasing the transparency of scientific assessments. WoE provides a systematic way to assess and integrate available scientific evidence addressing an assessment question.
41. The term WoE is often used colloquially to emphasize that all the evidence, confirmatory and/or contradictory, was taken into account in reaching a conclusion on an assessment question. The approaches and methods used in conducting such ‘non-formalised’, inherent, weighing of the evidence are mostly not spelled out However WHO’s (2009) definition of WoE is: “a process in which all of the evidence considered relevant for a risk assessment is evaluated and weighted”.
42. In conducting a transparent weight of evidence assessment, the following is required:
- A systematic, transparent way of collecting, assembling, and synthesizing the overall evidence.
- Systematic review methodologies to the extent possible.
- Clear qualitative and quantitative evidence is required.
Clear description of the approaches to weighing and assembling the evidence.
- Clear reporting of data and methodologies.
- Considerations of data quality and assessment of causal relationships.
43. Working definitions were then discussed. Weight of evidence is the extent to which evidence supports possible answers to a scientific question. This is reached by assessment of the balance of all evidence and can be expressed qualitatively or quantitatively. The evidence is systematically assessed in such a way that individual pieces of evidence (distinct element of information) are assembled into lines of evidence (a set of evidence of similar type). At each step, the evidence is weighed against relevance, reliability and, at higher tiers of integration, consistency.
44. WoE assessment for scientific assessment as a 3-step process was then explained (Figure 2). The question to be addressed by each weight of evidence assessment is defined by problem formulation, which is a step preceding the scientific assessment as a whole. This consists of defining the question(s) for the assessment and identifying which questions are required for the WoE assessment. WoE steps are assembling, weighing and integrating the evidence. The output of WoE assessment feeds either directly or indirectly into the overall conclusion of the scientific assessment. Although WoE assessment itself addresses some of the uncertainty affecting the scientific assessment, a separate step of uncertainty analysis is still needed to take account of any other uncertainties affecting the overall assessment.
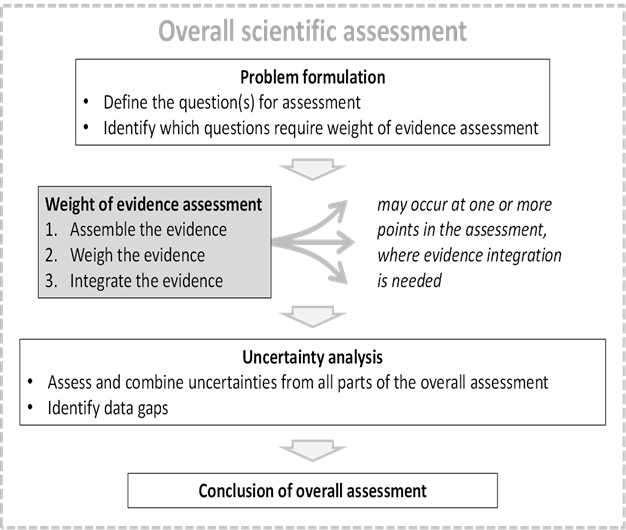
This flow chart is made up of a dotted outline text box. Inside the box are 4 text boxes with directional arrows. The interior text boxes contain a heading and bullet points. Diagrammatic illustration of weight of evidence assessment as a 3-step process which may occur at one or more points in the course of a scientific assessment.
Figure 2. Diagrammatic illustration of weight of evidence assessment as a 3-step process which may occur at one or more points in the course of a scientific assessment.
45. The relationship of relevance (including biological relevance), reliability and consistency are the three basic steps of WoE assessment.
46. Reliability is the extent to which the information comprising a piece or line of evidence is correct, i.e., how closely it represents the quantity, characteristic or event that it refers to. This includes both accuracy (degree of systematic error or bias) and precision (degree of random error).
47. Relevance is the contribution a piece or line of evidence would make to answer a specified question, if the information comprising the evidence was fully reliable. In other words, how close is the quantity, characteristic or event that the evidence represents to the quantity, characteristic or event that is required in the assessment. This includes biological relevance (EFSA, 2017) as well as relevance based on other considerations, e.g., temporal, spatial, chemical, etc.
48. Consistency is the extent to which the contributions of different pieces or lines of evidence to answering the specified question are compatible.
49. It was stated that there are no prescriptive approaches to WoE so soft titles were used. For example: Listing evidence, best professional judgement, causal criteria, rating, and quantification. Criteria have been defined to facilitate the choice of the WoE method(s) to be applied. These include the availability of guidance, the expertise needed, the ease of understanding for the non-specialist, the time needed, as well as the transparency and reproducibility of the methods, and aspects of variability and uncertainty.
50. Then the speaker discussed the classification of approaches adapted from Linkov et al 2009.
51. Listing evidence:
- Presentation of individual lines of evidence without attempt at integration.
- Best professional judgement: Qualitative integration of multiple lines of evidence.
- Causal criteria: A criteria-based methodology for determining cause and effect relationships.
- Rating: assess and integrate based on several factors, often derived from the Bradford-Hill considerations (e.g. International Agency for Research on Cancer (IARC), Occupational Safety and Health Administration (OSHA), Office of Health Assessment and Translation (OHAT).
- Quantification: Various statistical methods including meta-analysis.
52. Overall, the steps of a weight of evidence approach are the following:
- Assemble the evidence: Identify potentially relevant evidence (consider data gaps); select the evidence to be included; group into lines of evidence.
- Weigh the evidence: assess reliability, relevance and consistency; decide on method(s) for weighing and integrating the evidence (qualitative, quantitative, several); apply chosen methods and summarize results in a form allowing integration.
- Integrate the evidence: Consider the conceptual model for evidence integration to be applied (e.g. combining evidence with differing weight); assess consistency of the evidence (e.g. compatibility of lines of evidence); apply methods chosen for integration; develop conclusion for WoE assessment (summary of results, the way the conclusion will be expressed; and procedure for expert judgement if applied).
- Uncertainty and influence assessment.
- Potential for iterative refinement.
53. Reporting the outcome of a WoE assessment should cover all relevant information, in particular the approach to collecting and assembling the data, the type of data selected, the lines of evidence identified, and the method(s) used for evidence integration, Finally, uncertainty and variability issues should be addressed.
Benchmark Dose Modelling
54. Professor Þórhallur (Thor) Ingi Halldórsson presented on benchmark dose (BMD) modelling and the EFSA Guidance on the use of the benchmark dose approach in risk assessment.
55. BMD modelling in simple terms is two-dimensional curve fitting. A Benchmark Response (BMR) is defined as corresponding to a low but measurable change in response reflecting adversity. The lower confidence limit of the estimated change is then used as a reference point (RP).
56. There are multiple advantages in using a reference point derived using the results from the whole experiment (dose-response) instead of a simple pair-wise comparison (as used to determine the no observed adverse effect level (NOAEL).
57. EFSA has updated its guidance on BMD modelling over the years, with the first guidance being published in 2009, then updates in 2015 and 2022, respectively. In the 2009 guidance, the aim was to assess "the utility of the benchmark dose (BMD) approach, as an alternative to the traditionally used no observed adverse effect level (NOAEL) approach”. The conclusion was the BMD approach is of value…“in situations where the identification of a NOAEL is uncertain; when providing a reference point for margin of exposure (MOE) and a dose-response assessment using epidemiological data”.
58. In 2015, EFSA updated their guidance. It provided much more information on how to apply the BMD approach (excluding human studies). Model averaging is recommended as the preferred method of estimating the BMD and its confidence interval. The use of BMD modelling was not only recommended but the use of the NOAEL was strongly discouraged.
59. The speaker then discussed the BMD approach and the use of the NOAEL, picking up the main points from EFSA, 2015 as follows: “the benchmark dose approach is a scientifically more advanced method compared to the NOAEL approach for deriving a Reference Point (RP)”; “BMDL takes into account the statistical limitations of the data better than the NOAEL”; The Scientific Committee “strongly recommends that the BMD approach is used for the determination of the RPs for establishing HBGVs and for calculating MOEs”.
60. In 2022, EFSA updated its guidance following a workshop organised by EFSA in March 2017 to discuss commonalities and divergences in the various approaches for BMD analysis worldwide, and the update of Chapter 5 on dose response assessment of WHO/IPCS Environmental Health Criteria 240 (WHO, 2020). The Scientific Committee decided to again update its guidance in order to align the content of the document and harmonise further EFSA’s approach with those of its partners.
61. The main changes are a shift from a frequentist to the Bayesian approach, a more extended set of models for model averaging; the normal distribution is now “finally” included to harmonize with other agencies, and it is more open to the use of alternative solutions when limited dose response information can be extracted. It was noted that The United States Environmental Protection Agency (EPA) have 10 different empirical conditions to flag “questionable” model outputs.
62. It is important to note that the NOAEL is still a method available as a last resort.
63. In summary, the current EFSA guidance on BMD methodology has been aligned with other agencies (US-EPA, WHO); the methodology and its computational platform has been implemented “close to perfection”, however there is always a “but”. BMD modelling may not always be the most sensible solution, but it should be one of several options in the toolbox. This conclusion is supported by “a patchwork of conditions” found in the guidance to eliminate exceptions.
64. The series of guidance updates by EFSA have resulted in relatively well thought through guidance on how to perform BMD modelling. It is currently difficult to see how the modelling aspect can be improved further, although the computational platform could be improved, and the underlying assumptions could be made more transparent.
65. EFSA recommends the use of BMD modelling by default and strongly discourages the use of the NOAEL approach. However, models cannot extract more information than the underlying data contains.
66. Experiments with few dose groups (n=3-5) and few animals may not always contain the level of information that the risk assessors need. Future guidance should allow for alternative approaches giving more weight to common sense along with statistical considerations.
Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH)
67. Mr Keith Bailey talked about REACH in the regulatory context and some of the thing’s COT might want to take into account in its guidance.
68. The speaker first introduced REACH Article 1. The purpose of this regulation is to ensure a high level of protection of human health and the environment, including the promotion of alternative methods for assessment of hazards of substances, as well as the free circulation of substances while enhancing competitiveness and innovation.
69. This regulation is based on the principle that it is for manufacturers, importers and downstream users to ensure that they manufacture, place on the market or use such substances that do not adversely affect human health or the environment. Its provisions are underpinned by the precautionary principle.
70. It is important to note that this is the paradigm shift of REACH; responsibilities fall on both industry and the regulator. Pre-REACH, when new substances were put on the market it was the responsibility of industry to do the risk assessment before those new products got onto the market. For existing substances, the regulator did the risk assessment. Now everything is put on the same footing, and the responsibility primarily lies with industry. Therefore, it is important to flag that guidance needs to work for industry not just regulators.
71. REACH primarily works at the level of the individual substance but industry and regulator risk assessment can be carried out based on groups of chemicals. Industry may need to consider the chemicals they are producing as a group and think about how to do so. The regulator can view it different ways, grouping by substance or by similarity of use. Some examples include CMRs (CMRs are substances that are carcinogenic, mutagenic, or toxic to reproduction), textiles, tattoo inks.
72. Identification of hazard and assessment and management of risk are threads that run throughout REACH.
73. Next the speaker discussed the responsibility of the manufacturer / importer in chemical safety assessment, which applies only to substances to be put on the market at a level above specified tonnes per year.
74. The purpose is for manufacturers and importers to assess and document that the risks arising from the substance they manufacture, or import are adequately controlled during manufacture and their own use(s) and that others further down the supply chain can adequately control the risks.
75. REACH is looking at individual substances as compared to regulatory regimes which start with an end product. A lot of information that REACH is looking for is designed to answer questions regardless of what the end product is.
76. Step 1: hazard assessment (human health, phys-chem, environmental, Persistent, Bioaccumulative and Toxic (PBT) / very Persistent and very Bio-accumulative (vPvB).
Step 2: where the hazard assessment identifies specified hazard classes or assesses the chemical as PBT/vPvB it must be followed by exposure assessment (exposure scenarios & exposure estimation) followed by risk characterisation, which considers the human populations and the environmental spheres for which exposure to the substance is known or reasonably foreseeable and the overall environmental risk.
77. The chemical safety assessment is written up in the Chemical Safety Report and submitted as part of the registration dossier.
78. It is for manufacturers, importers, and downstream users to ensure that they manufacture, place on the market or use such substances that do not adversely affect human health or the environment.
79. For the vast majority of chemicals, the duty to carry out risk assessment lies only with industry, which covers roughly about 5,000 substances.
80. The role of the regulator: Health and Safety Executive (HSE) compliance-check a proportion of registration dossiers, which is around 100; registration compliance-check a minimum of 20%, checking the quality of the work, but they do not re-do the risk assessment.
81. Processes where there is an element of regulator risk assessment: Substance evaluation, prioritisation for authorisation, restriction, are there any gaps? if so, the regulator would then require more information from industry. Risk management option analysis (non-statutory), such as Classification, Labelling and Packaging is also required.
82. Granting of authorisations is back to regulatory scrutiny of industry applications.
83. REACH registration is based on standard test methods such as Organization for Economic Cooperation and Development (OECD) Test Methods, which would be added into REACH Annexes. It is important to distinguish between the standard test methods and what is necessary and appropriate for a particular chemical.
84. REACH Annexes have seen changes to standard test methods for some endpoints with replacement / reduction of animal testing. REACH has seen a lot of refinement in animal testing over the years.
85. Therefore, it is a case-by-case consideration: endpoint-specific or general waivers, last resort principle, testing proposals, regulatory requirements from compliance checking or substance evaluation and even using the term weight of evidence. One would have to justify why such a chosen methodology has been used. However, who demonstrates efficacy in these cases? Standard test method OECD validation.
86. There will be challenges in the resources available to assess the efficacy of any NAMs proposed.
Roundtable Discussions from Session 1
What could work better?
87. It was considered that EFSA may use multiple uncertainty factors, which can be problematic and difficult to justify. Therefore, this should be something to be avoided, if possible, in the new COT guidance, which should make every effort to replace these uncertainty factors with data-derived factors. However, avoidance of uncertainty factors will require additional knowledge than perhaps we currently have.
88. More transparency is needed in how the COT performs its risk assessments, possibly necessitating a way to formalise its way of working, and then integrating this into a compatible guidance that is already available.
89. The COT needs an overarching guideline/guidance document but there is reluctance to develop its own framework; it would be preferable to adopt existing guidance. However, there is uncertainty on which would be the most applicable and / or if possible, whose choice and what criteria need to be fulfilled in order to determine which sections would be appropriate to select.
90. There needs to be a clear distinction between who is responsible for what in the different sister committees and SACs involved in chemical risk assessment more generally.
91. The guidance should allow for the use of grouping and weight of evidence. A brief discussion was had about what sources should contribute to WoE, leading on from the earlier presentation. The points made were that the evidence used must be based on scientific fact, and sources of opinion, such as social media, would not be appropriate. However, it was accepted that evidence can arise from less typical sources and still contribute towards WoE.
92. Resourcing will be key to use these new tools. Therefore, there should be dialogue between the regulator and industry to encourage harmonization.
How does COT guidance fit with other guidance?
93. It was suggested there should be a review of what other guidance is currently available that represents good practice, before determining what could be learnt from this guidance and then consider what would be appropriate for Great Britain (GB) specifically. It was noted there is a lot of similarity between guidance from different bodies, therefore by reviewing all of the guidance available, the COT could decide which aspects would and would not work for GB. It was indicated that harmonisation across guidance would be preferable and while the implications of being too divergent from the European Union should be considered, this chance to review guidance from a GB perspective should be seen as an opportunity for GB to identify gaps and become a leader from a regulatory and industry point of view.
94. Harmonisation and lessons learnt from this other guidance could be applied to any GB guidance.
95. The guidance could be re-written for applicants for the various regulated product areas and subsequent committees and JEGs. It was stated that re-writing of the guidance to make it more relevant and bespoke for GB applications was a lower priority and currently committees do not have the time to discuss this whilst there are still large numbers of dossiers to be evaluated.
What other guidance is out there?
96. Guidance’s suggested from which GB could learn include Health Canada, European Chemicals Agency (ECHA), WHO and General Data Protection Regulation (GDPR).
97. It was agreed that information was readily available and easy to access, meaning individuals could select appropriate guidance to use, based on their need at the current time.
98. It was questioned whether the other guidance would be applicable only to the COT, with agreement that any new guidance should work for the COT sister committees; Committee on Mutagenicity (COM) and Committee on Carcinogenicity (COC) as well as SACs advising other government departments.
Can priorities be identified?
99. Concerns were raised that should the GB guidance differ too far from guidance from other existing bodies industry would decide to work to EFSA or USA guidance over GB. However, it was considered GB should use good practice and be seen as a leader in the field. It was added that, from a scientific point of view, the principles behind the guidance are (and should be) similar across existing bodies, and this exercise was more about what could be added to GB guidance so that it was tailored towards what works best for GB specifically. Emphasis was placed on the GB guidance working in the most beneficial way to GB.
100. It was agreed that GB should liaise and build relationships with other national agencies, especially countries with their own guidance such as Canada and Australia. It was highlighted that Health Canada in particular had done a lot of work on AOPs which may be useful for GB guidance.
101. Members asked whether more quantitative methods be incorporated, and how could dose-response be incorporated.
102. It was stated that a useful development would be to produce a skeleton of how assessments could be done at the COT.
103. It was stated that it would be important to define the scope of the guidance because the remit of COT is so broad, so any new guidance may also have implications for other government departments such as HSE and the Department for Environment, Food & Rural Affairs (DEFRA). Therefore, the approach should be harmonised across these different GB agencies so that any advice can be directly compared. In light of this, it was further queried whether this should be a COT decision at all and if so, how far would COT’s remit extend. It was suggested that, given the number of departments and committees this guidance could affect, that perhaps this was beyond the scope of COT and that perhaps this task sits above one individual department/agency. Concerns were raised that a lot of resource would be required in order to reorganise the system, and an alternative government body taking responsibility for the entire system restructure might be better.
104. There was discussion on how NAMs may be incorporated into the guidance. Their relevance for human toxicity, and if/how wholesale replacement of more traditional approaches could be affected in practice. NAMs, for instance, can throw up unknown areas of concern, and potentially identify non-apical endpoints that are difficult to interpret. Such uncertainty can be a discouraging factor for industry to use NAMs in regulated product applications and it was suggested that in the SACs’ dossier evaluations a less stringent evaluation could be given to such studies. It was stated that animal studies will not be removed entirely, but animal data should be used better. A lot of important NAMs approaches still rely on animal data but do so with new approaches. Indeed, it was concluded that no one size fits all, and relationships or correspondences between NAMs outcomes and those in more traditional toxicology studies, epidemiological studies, and biomarker studies should be identified and pursued (e.g., using NAMs to fill the data gaps from the animal studies). There is a need to move towards more AOP driven approaches. It was stated that toxicological evaluations relating to NAMs are in a different place than for example 10 years ago. Although academics may have been overly optimistic about the prospects of NAMs, there has been a shift and NAMs are currently functioning in more appropriate areas.
Other topics raised
105. The question was raised of whether regulation should follow science rather than other way around. Evolving regulations gradually over time is a good approach but could be confusing and require too much work. A descriptive remit may be beneficial but could also be too specific. Overall, risk assessments and guidance should be science led, not stipulate specific test requirements, but adapt as the science changes.
106. It was discussed how the Advisory Committee on Novel Foods and Processes (ACNFP) are still following the EFSA guidelines when evaluating novel foods, and this may not always be the best way forward. For instance, these are products intended for direct consumption and in animal studies overt toxicity is rarely seen, standard 90-day studies use large numbers of animals, and some studies that are submitted are of a low quality (e.g., university final year PhD studies that are not fit for purpose). It was questioned if this comprehensive toxicology evaluation approach should be actively discouraged? An important issue that was raised in this respect, and again in session 3, was the distinctions between different chemicals based on their intended (and unintended) uses, and if/how their evaluation should be reflected in guidance. With respect to REACH, for instance, it was suggested that this approach is fundamentally needs-based and not hazard-based; it is based around volumes of chemicals.
107. Regarding threshold, members discussed that there may not be a practical threshold, but that for genotoxicity specifically there must be a threshold. It was stated that it can be difficult to identify such thresholds.
108. The BMD approach is a considerable improvement on what had been done before. This was then linked back to genotoxicity where it was stated that there are no suitable tests to allow the dose response to be adequately characterised and that it would not be possible to quantify the risk.
Session 2: What we need to improve?
Building regulations
109. Dame Judith Hackitt presented on building a safer future: changing an industry culture; the need for Leadership, Pride, Professional Competence and an effective Regulatory Framework.
110. Although Dame Judith’s talk covered a very different area to that of chemical risk assessment, it was noted that it is useful to get someone from outside to look in and give an objective assessment at a fairly high level at whether a system is working.
111. The speaker then gave a timeline of the Grenfell Inquiry. In July 2017, a public inquiry was announced, and it is still ongoing. In August 2017, an independent Review of Building Regulations and Fire Safety (the briefing being What went wrong? And would this affect other areas?) started and the final report was published in May 2018. In July 2018, an Industry Safety Steering Group was established and in late 2019 a new Building Safety Regulator was announced. In July 2020, a draft Building Safety Bill was published for scrutiny; April 2022 Building Safety Act received Royal Assent. The detailed publication is due next year.
112. The speaker then discussed that they had aimed to ensure that the process gets followed through and delivered in the right way. The need for continuity is key and clarity of purpose in the convoluted process of getting a new regulatory system up and running and delivering the right culture. The regulatory system is there for a purpose. It is there to change behaviours, to deliver the right behaviours and to deliver the right outcomes. If you focus only on regulation and don’t think of the people’s behaviours that are being driven by that regulation, we don’t focus on the right things.
113. We need to think about transparency in the information being submitted or omitted.
114. The process of the system was already complicated but also many other things happened in the world such as Brexit, Covid 19, massively increased awareness of climate change, political uncertainty which complicated the process even more and made priorities change over time. Therefore, when you want to make change in a system you need a champion to maintain focus and drive.
115. The speaker discussed how she reviewed the system, which showed that it was broken, not fit for purpose, and needed to be fixed. Especially, she focused on how everything is siloed and not stitched together. The speaker then elaborated on the diagnoses of the problem.
116. It was found that the current regulatory system for ensuring fire safety in high rise and complex buildings is weak and ineffective with little or no enforcement; therefore, a new regulator is needed. The industry behaviour is characterised as a “race to the bottom” with significant evidence of gaming the system. Conflicts of interest abound. The design, change management and record keeping is poor, both during construction, occupation, and refurbishment. In general, the experts, as well as the residents, are not listened to. The culture of the whole construction and built environment industry must change. Product testing, marketing, labelling and approval processes are flawed, unreliable and behind the times; therefore, a new regulator is needed. There is a general lack of assured competence and quality assurance across the sector. There is an unspoken knowledge about the extent of shortcuts being taken without real assessment of potential consequences.
117. Only by standing back did these things became clear. We need to put in place a new regime underpinned by a set of key principles; a systems-based approach to both regulations and to buildings. Competence, accountability, and responsibility should be at the heart of the system and have the right players in the right system. A culture change with positive incentives for good building practices and those who are willing to stand up and be counted for doing the right thing; and punitive sanctions for those who continue to try to game the system. We need a risk-based, proportionate regulatory framework and an outcomes-based framework to encourage real ownership and accountability. This would be overseen by the regulator with those undertaking building work needing to demonstrate they know what they have and are doing all they can to manage and reduce risk. Focus on delivering quality buildings which are safe and feel safe to live in whether single dwelling or multi-occupancy. Genuine engagement with and concern for residents and rebuilding of trust and reputation.
118. The speaker started to map out how the system was supposed to work. It was complex and it was too complicated for it to work properly, and it provided lots of opportunities to bypass the system. Therefore, a new map was devised that simplified the process which is what the regulatory act is designed to do. We are now moving to a system where responsibilities are clearer, and the driving culture changed. The role of regulation is clear, to hold industry to account including underpinning of 3rd party accreditation.
119. The links and running thread should be quality, sustainability, and resilience.
120. Since the report, there has been progress in driving culture change ahead of regulatory change.
121. Learnings over the last 4 years from the Industry Safety Steering Group, which is driving the needs from the right behaviours from industry, were then discussed. There is progress on all fronts and pace is accelerating now that regulation is in sight. However, too many are still waiting to be told what to do and asking for detail. Industry leadership is emerging but not fast enough and not enough of it. Insurance and financial markets are now alert/sensitised to concerns. All the players involved are contributing to getting the right answer. There are challenges of new build and existing stock, which are different. Voluntary Building a Safer Future Charter is an important development to drive change in the sector. Professional competence is fundamental, and it is not enough to know your own job/trade but you also need to understand its impact on system integrity overall.
122. We must really focus on what we are trying to achieve. The speaker stated that they were trying to make buildings that are fit for purpose, built to the right quality, and maintained at their quality throughout their use, not just at the point that they are finished.
123. A Safety Case regime for higher risk buildings makes people think about what might happen and how to manage consequences. Gateways at design and commissioning stage are needed and actual delivery on what these say.
124. It won’t be enough to say what you intend to build; it must be demonstrated what has been built based on evidence and proof. The supply chain needs to be ready and able to provide data and performance accreditation.
125. Collaboration and data sharing are key so that every single person understands their role in that system and delivers on the overall picture.
126. This can include standardised system(s), performance testing, less substitution, advice on use and application from competent persons, regular integrity checks and “product stewardship”.
127. When you change any regulatory system or review it, it has ripple effects and can drive a culture change. There is always more change to come, the regulatory system is dynamic.
128. This is a once in a generation opportunity to leave the race to the bottom behind and change industry practice for good.
129. An integrated systems-based approach to creating the whole regulatory framework is essential.
Evolving our assessment
130. Dr David Gott presented on a golden thread through past, present, and future.
131. The speaker started with the regulatory toxicology tragedy of thalidomide and how that enhanced and checked our thinking for 50 years. We need to move on from this and now start thinking differently about our approach to safety assessment.
132. The risk assessment paradigm doesn’t exist in isolation. We need to think how to use the best science that will tell you the best answer and then you take other factors into consideration towards the risk assessment process.
133. The speaker then used nanomaterials as an example of evolving guidance.
134. Nanomaterials have been around for a number of years, but we were not specifically considering them. It wasn’t until a number of reports on their potential uses and risks emerged at the start of millennium that the scientific community investigated this. Thereafter, a 2005 joint COT/COC/COM statement outlined principles for evaluation, issues, and major uncertainties. They acknowledged that a lot of problems couldn’t be answered. This then triggered a lot of research. EFSA started its journey on developing guidance, which evolved over a 15-year journey using fundamental principles. EFSA then illustrated how they were going to apply the principles and outlined detailed approaches to evaluate and test nanomaterials.
135. We live through change and will continue to do so. The principles for assessment should be the same, whether we assess something that is added deliberately or is something that is already there, the principles and the science behind them stay the same. The guidance should describe how we use the available information to estimate risk. On the other hand, NAMs should use the minimum data needed to satisfy us on the lack of any meaningful risk.
136. The speaker then discussed the toxicological paradigm and how we should use exposure more and how biological effective dose is still mainly unknown. Population dose response dynamics will be key to protecting the right people because we are all different but normally, we aim for protecting the most sensitive population.
137. The Joint FAO/WHO Expert Committee on Food Additives (JECFA) risk assessment approach was explained. They had the same general principles and methods for all chemical risk assessments. They have published in their reports since the mid-1950s and recommended reviewing the validity of the evaluation procedures then in place in the early to mid-1980s based on good science at the time.
138. The International Programme on Chemical Safety (IPCS) published Environmental Health Criteria monographs (EHCs) on Principles for the Safety Assessment of Food Additives and Contaminants in Food, EHC 70 (IPCS, 1987), and Principles for the Toxicological Assessment of Pesticide Residues in Food, EHC 104 (IPCS, 1990). Essentially, these told you how to interpret the data. Around 2000, IPCS recognised there has been significant advances in chemical analysis, toxicological assessment, and risk assessment procedures.
139. They took particular note of two reviews. of the IPCS Harmonization Project and the Food Safety in Europe project of the European Commission (Barlow et al., 2002; Renwick et al., 2003), which were used to inform the development of a revised monograph EHC 240: Principles for Risk Assessment of Chemicals in Food, which describes general principles and methods for the risk assessment of additives, contaminants, pesticide residues, veterinary drug residues and natural constituents in food.
140. When it came to guidance and what data we might need, the traditional guidance of the Scientific Advisory Committees (SAC) was derived from the 1960s approaches to pharmaceuticals. They emphasised on trying not to miss anything because of previous experiences such as with thalidomide. Therefore, they identified the need for specific toxicological studies and appeared to require them, which at the time meant studies in two species.
141. Then there was an opportunity to reboot, which started with a review of existing guidance and what we now knew. Discussions were on what data were used and what was needed for a decision, recognizing that one size does not fit all. There was a desire to learn from the past and be ready for the future evolving a much more scientific approach.
142. This evolution turned out to be what is now known as a tiered approach (for some categories of chemical, such as food additives), which is about thinking about your data rather than just generating data. Thinking about it on a case-by-case basis. It is about the physicochemical data on the compound, the toxicity data on structurally related compounds and any available information on structure activity relationships. Animal testing was in line with the 3R strategies. This is inherent in the rationale that results of studies at higher tiers will in principle supersede results at lower tiers which was an advantage. The applicants were able to identify relevant data needs more readily, and it allowed adequate assessment of risks to humans and strengthened the scientific basis for the assessment.
143. The tiered approach consists of 3 tiers: testing requirements, key issues and triggers are described. The minimal dataset applicable to all compounds has been developed under Tier 1; Tier 2 testing will be required for compounds which are absorbed, demonstrate toxicity or genotoxicity in Tier 1 and Tier 3 testing should be performed on a case-by-case basis taking into consideration all the available data, to characterise specific endpoints needing further investigation from findings in Tier 2 tests.
144. The speaker accepted that it was not perfect and that it had errors as well as flaws as it required compromise in what people were prepared to contemplate. However, it does balance the data requirements against the risk. It initially uses fewer complex tests to obtain hazard data and evaluated to determine if they are sufficient data for risk assessment or, if not, to design studies at higher tier.
145. Some of the unintended challenges were that they worried about limited experience in what panels would consider sufficient and struggled with the emphasis on integrating information not just test results. Scientists generally welcomed the tiered approach and its emphasis on good science. Others tried to over interpret it and the meaning of words chosen by consensus (and able to be understood by non-native speakers).
146. It was an achievement as it did what is set out to do which was to lower the amount of data required and accept animal use if it was used properly.
147. Integrating the evidence is absolutely key, and the critical part of the risk assessment. We need to identify all the potential hazards and determine if they need characterising and whether the toxicological data internally is consistent with our wider knowledge. It is important to note that every experiment makes design compromises and possibly uses small numbers and high doses so that there is a need to think about possible changes in kinetics.
148. We need to think about using all the data sources such as human studies, animal studies and in vitro studies. These will contribute to dose response information on what the compound does to the body (dynamics) and what the body does to the compound (kinetics), noting that these relationships use models, which may or may not reflect reality. When providing the risk assessment, we have to be honest to risk managers in that we may not know all the answers and point out what is uncertain.
149. The speaker then discussed personal concluding thoughts on the next evolution and future challenges. There needs to be increased emphasis on toxicokinetic information to allow evaluation of internal dose and reduce reliance on default uncertainty factors. There needs to be greater emphasis on integrating and including information from various sources. More thought on relevance of changes for adverse effects in human is needed. We need to start to think about intermediate end points and how to incorporate and use them. There need to be indications of when we can rely on less data or testing.
150. Some of the challenges will be how to incorporate NAMs and omics technologies into our framework. There are lots of roadmaps, perhaps maybe we should be radical and just do it. What should we do about the unknown unknowns as well as uncertainty factors. How much validation will we require. Biology and medicine have a long history of serendipitous findings and good ideas that were based on imperfect knowledge. Do we need to revisit the acceptable daily intake (ADI)/ tolerable daily intake (TDI) concept, as well as revisiting what is an appreciable health risk. Should we be pressing risk managers to ask what is dangerous rather than tell me a safe intake. We need an estimate of the amount of a substance in air, food or drinking water that can be taken in daily over a lifetime without appreciable health risk.
151. Values are not always good estimates of the harmfulness of chemicals.
152. Deciding on what level of risk is acceptable is down to society. We will need to inform this debate with reality and accept what is unachievable. We will need to acknowledge we cannot change some events which will happen to all. Be clear in every choice and decision will have consequences; known and unknown. We know a lot but not everything and in hindsight some things will be wrong due to that. Everyone is an individual who makes choices and it’s important to remember that in future. We might prolong life and quality of life better but there are some things which are beyond our control.
Roundtable Discussions from Session 2
Gaps identified and recommendations for improvement
153. Guidance: The scope of the current guidance/guidelines should be explored and defined. The guidance must emphasize limitations of extrapolation of raw data to observed and expected results. There is a need for a systems-based approach. Tiered testing could be incorporated such as toxicogenomics in vitro, which is considered a conservative approach and appropriate for low tier screening.
154. Frameworks: It was questioned whether a different framework is needed to distinguish between those things that are purposefully added to food and those which occur as contaminants. For example, are different datasets required for exposure assessments? It was discussed that exposure assessments for COT papers tend to be very thorough and should maintain this level of detail and perhaps extend it? The idea of storing data from COT exposure assessments in a centralised accessible format was suggested. Each COT paper contains a lot of information, and ideally risk assessors should be able to easily access and reuse this data as appropriate. It was proposed to have an overarching chemicals framework that would link to various committees and advisory groups for specific categories of chemical.
155. NAMs: The need for a deep-dive analysis on where NAMs have been applied and what indicators could be used to support and/or supplement what may be needed for the guidance (i.e. to prevent loopholes). There is a need to be able to interpret data generated by NAMs (e.g. computational models) as a useful resource for hazard identification. but where to draw the line? Will need to understand the method as well as the limitations of the outputs. We need to ensure that a bigger picture of where NAMs fit in to the regulatory space is kept in mind. It was stated that some people’s experience of NAMs is that they’re difficult to interpret. It is hard to pinpoint specific requirements for a NAMs study. It was also discussed that a basic study data set should not be stipulated. The data should enable hazard characterisation, exposure assessment, and risk characterisation and the guidance should not be a set of check boxes. A key question was whether mechanistic links to adversity are needed? However, it was raised again that defining ‘adversity’ is a key challenge that revolves around use of language in different contexts. The meaning behind the name (New Approach Methodologies) varies, and one definition is not limited to methodologies that are purely in vitro or in silico, but instead takes account of any novel approach. Hence, some NAMs may include in vivo methodologies, for example, you could take blood samples to assess the kinetics (this approach is a compromise) or may include reporter genes for specific stress responses. Testing hypotheses may not be possible without the use of a gold standard method so it may not be possible to fully move away from animal studies (where they are the gold standard) for the foreseeable future.
156. SACs: There is a need to strengthen the link between the three committees (and Joint Expert Groups) to ensure a consistent and harmonised approach. It may be necessary to do this across all Committees that provide advice on chemical risk to the government. Regular transparent communication between each of their activities was also identified as a gap. Noted that there is a limited pool and capacity to recruit new experts.
157. Does the COT need to present a narrative review of all studies, or might a tabular and/or graphical representation be more useful, easier to digest, and informative? Narrative review can sometimes be overwhelming, or conceal the most useful aspects of the paper, the COT therefore needs to work to draw these aspects out, summarise, and present them. It is important to draw a balance between providing systematic reviews and narrative reviews in COT discussion papers and statements. The limits of accessibility requirements were mentioned, and that they may potentially preclude such practices although noted that extensive tables and graphs are fine if accessible alternatives are provided. The suggestion of segmenting papers into more and less accessible sections was suggested.
158. Judging the quality of a published paper is not a tick box exercise. There are more problematic practices in academic publishing, like intentional manipulation of data or the use of paper mills that produce papers that look legitimate, but which may use fabricated data.
159. The Regulators: It is fundamental to capture institutional wisdom. When compared to US departments (e.g. EPA, National Toxicology Program (NTP) and National Institute of Environmental Health Sciences (NIEHS)) there seems to be a good link between them, where responsive research is possible There doesn’t seem to be anything similar in the UK, though the participants were aware of UK Research and Innovation (UKRI). There were challenges of time and resources on how to review large datasets. We will need to develop ways/tools on how to handle large volumes of data (especially in the case of Regulated Products, Trade assessments), and specify available toolkits. There is a need for more “quality” resources for risk assessors, to supervise and lead. Assessors’ need training in how to assess and review all the different types of data, therefore build that into the time frame. An overarching issue is how to deal with different interpretations of the data, so we need to develop a standardised way, but this may become a tick box approach. There is a gap between integration of sustainability, industry, safety, and regulatory aspects of GB chemical regulations/strategies. Purely academic papers don’t necessarily inform work on regulated products, as a lot of these papers omit key information that is not necessary for academic publications. They have different objectives than informing risk assessment so this will be a future challenge to note.
160. Assessment: This discussion raised issues related to HBGVs and their utility/application, the differing interpretations of language in different fields, and the way data presented to the COT may be archived and databased for future use in assessments. There was also discussion about acute exposure vs. chronic exposures and the requisite datasets, and risk assessment for deliberate consumption versus contaminants.
161. The discussion began by considering whether lifetime exposure is a relevant index. For instance, sometimes the ADI is the only tool available, but its use can give rise to overly conservative assessments. This led into a discussion about language, and the meaning of ‘dangerous’ vs ‘safe’, and although a value can be set that indicates no appreciable risk, it does not indicate at what level above this there’s a health concern. It is impossible to exclude that something might be happening. For instance, cadmium exposure is never ‘safe’, but reducing exposures is not always possible. Safety is not a binary scale, with safe and unsafe being the only options, but how this is communicated is difficult.
162. The GB risk assessment system needs a gradual evolution. It was stated that it is currently functioning well, but perhaps clunky due to age and reliance on animal testing.
163. Key issues around uncertainties relating to mixtures and interactions are important to consider going forward, particularly on how chemicals interact and how these interactions can be integrated into risk assessments. It was also stated in this context that getting the problem formulation correct is a crucial starting point.
Session 3: How to achieve?
What is required by Retained EU Law (Revocation and Reform) REUL?
164. Dr Erica Pufall introduced a session on Retained EU law (REUL). Retained EU law is a legal term introduced into UK law under the European Union (Withdrawal) Act 2018. It is a tailored legal concept capturing EU-derived laws, rights and principles retained and preserved in UK law for legal continuity after the transitional arrangements under the Withdrawal Agreement.
165. REUL is all the EU law that was incorporated into domestic UK legislation when the UK exited the EU; all the laws that were needed were copied into UK law, and this became ‘Retained EU Law’. REUL makes up >90% of UK food law.
166. The Retained EU Law (Revocation and Reform) Bill (“REUL Bill”) was introduced into the House of Commons on 22 September 2022.
167. This Bill allows for the revocation or amendment of REUL and removes the special features REUL has in the UK legal systems making it significantly easier to undertake reform in the longer term.
168. In Northen Ireland, under the Northern Ireland Protocol, much of EU food and feed law is directly applicable, rather than being REUL. The FSA are working through any implications of the Bill for Northern Ireland food and feed law.
169. The REUL Bill proposed that what were previously EU laws would be removed from the statute books before 31 December 2023, unless departments and the Devolved Administrations took steps to move them into domestic legislation. If departments did not act to transfer REUL into domestic legislation, it would fall off the statute book at ‘sunset’ of 31 December 2023.
170. On the 10th of May, the UK government tabled an amendment to the Bill to replace the sunset clause with a revocation schedule that lists the REUL to be revoked/sunsetted by departments. This effectively flips the Bill on its head, changing the default position from lapsing REUL to preserving it.
171. This schedule is a list of the retained EU laws the UK Government intends to revoke under REUL at the end of 2023. All other legislation not on the Schedule will automatically become domestic law.
172. The Revocation Schedule includes eight pieces of law the FSA is responsible for. The FSA has carefully examined these eight pieces of legislation and is confident that removing them will not impact on the safety or standards of UK food. Our list of REUL to be revoked can be found here: Retained EU Law to be sunset or revoked by 31 December 2023 | Food Standards Agency.
173. The speaker then discussed what REUL means for the FSA. Retained EU law covers a range of areas relating to food and feed safety. The FSA has long standing ambitions to reform the food and feed regulatory system and the REUL Bill presents a significant opportunity to undertake legislative change to support our wider ambitions for reform. We have ideas on where we can make significant improvements, which we are discussing with industry to ensure we understand and address potential concerns. For example, an area the FSA has already committed to reviewing is the regulatory framework for Regulated Products, including Novel Foods. This will include how the process for approving novel foods could be updated, to create a transparent and effective system that is the best in the world for innovators, investors and consumers and encourages safe innovation in the sustainable protein sector.
174. The FSA Board have agreed five principles to guide our approach to REUL:
- Protecting public health, food safety and standards: we should not make changes which reduce the safety or standards of food produced or eaten in the UK.
- Protecting consumer interests: we should not make changes which are detrimental to the wider consumer interest in relation to food (for example by making food harder to afford).
- Maintaining consumer and trading partner confidence: we should not make changes which are likely to reduce consumer or trading partner confidence in UK food, or which are inconsistent with our international trade agreements.
- Supporting innovation and growth: we should seek to make changes that support innovation, growth and the introduction of new technologies, (including innovation that could help to make food healthier or more sustainable) and remove unnecessary burdens on business.
- Managing divergence: we should, as far as possible, seek consistency of approach across GB, in line with common framework commitments. We will consider the impact of divergence on public health and safety and the wider impact for consumers or businesses of having differing rules across the UK.
HSE Pesticide NAMs Case Study
175. Dr Valerie Swaine presented on the experience with NAMs in Plant Protection Products (PPPs) toxicology assessments in GB (Great Britain).
176. The speaker stated that the HSE definition of NAMs is in silico, in chemico, in vitro and in vivo methods and a combination of these that can provide information on chemical hazards and risks, by replacing or reducing the use of animals.
177. The speaker then discussed PPPs as an example of chemical mixture to which NAMs are being applied. For authorisations of these products a standard “six-pack” of acute toxicology animal studies is required, however Article 62 of the PPP Regulation says: “Testing in vertebrates only where no other methods are available”. By applying Article 62, it is possible to meet the data requirements without performing animal testing. For acute oral, dermal and inhalation toxicity, HSE applies the calculation method for mixtures of the CLP (Classification, Labelling and Packaging) Regulation; for skin and eye irritation HSE encourages the use of in vitro OECD test methods and OECD IATAs; for skin sensitisation HSE recommends the use of the recently adopted in vitro OECD defined approaches; and for dermal absorption HSE requires the in vitro skin absorption (OECD 428) method.
178. HSE has also issued guidance to applicants on Meeting the requirements for toxicological information in applications for authorisation of Plant Protection Products under Regulations (EC) No 1107/2009 and (EC) No 284/2013 where these alternative approaches are explained.
179. The speaker then went into brief details for each of the examples.
180. For acute oral toxicity, classification of mixtures can be determined using the additivity formula of CLP by calculating the acute toxicity estimate (ATE) of a mixture. This is based on the available acute oral toxicity data on each component of the mixture and their respective concentration in the mixture.
181. For skin corrosion / irritation, the OECD Guidance Document on an Integrated Approach on Testing and Assessment (IATA) for Skin Corrosion and Irritation which was published in 2014 can be used.
182. The IATA provides a framework guiding users on testing and assessing skin irritation for:
- Classifications & Labelling decisions.
- Weight of evidence (WoE).
- Approach to testing.
- Further information requirements.
183. For eye damage / irritation, the first approach was published in 2017 and then updated in 2019. The eye irritation IATA framework provides similar guidance to that of the skin irritation IATA. However, at the time of publication in vitro methods could identify only chemicals that are classified as corrosive (Category 1 (H318)) or non-classified (non-irritant), whilst none could identify category 2 eye irritants, therefore a WoE assessment and occasionally an animal test was required. The OECD published last year a new eye irritation in vitro method and this one has been recommended as a full replacement of the in vivo test. It is called the SkinEthic™ Human Corneal Epithelium (HCE) Time-to-Toxicity (TTT) test. It is able to correctly identify chemicals (both substances and mixtures) in the 3 CLP classification categories. The Guidance Document No. 263 on the IATA can be consulted for further testing with other adequate in vitro tests, if deemed necessary.
184. For skin sensitisation the OECD published in 2021 a Guideline (No. 497) using Defined Approaches, which are based on in vitro, in chemico and in silico tools. Guideline 497 is the first internationally harmonised guideline to describe a non-animal approach that can be used to replace an animal test to identify skin sensitisers. Defined approaches are slightly different from IATAs as they define a set of information sources to derive a prediction without the need for expert judgment. Results from multiple information sources can be used together in defined approaches to achieve an equivalent or better predictive capacity than that of the animal tests to predict responses in humans. One example of a defined approach is based on a combination of in vitro/in chemico tests to address some of the key events of the AOP for skin sensitisation. This is then combined with a (Q)SAR prediction. A score is then obtained which allows the prediction of the skin sensitisation hazard potential and potency sub-categorisation of the substance/mixture according to the United Nations Globally Harmonized System (GHS) of Classification and Labelling of Chemicals.
185. The speaker then discussed HSE activities and application of NAMs to pesticide active substance assessments. HSE is currently working at international level to develop a waiver for the dog study. HSE is also working with the UK Committee on Carcinogenicity to develop a new testing strategy for the carcinogenicity assessment of chemicals which is currently based on two assays (one in rats and one in mice) using a large number of animals. HSE supports greater use of read-across between active substances, with possible augmentation obtained through the application of omics technology. In addition, HSE encourages an increased use of (Q)SAR for metabolites and impurities to fill data gaps. Another application of NAMs is the use of the Threshold of Toxicological Concern (TTC) Cramer Class III value in the relevance assessment of groundwater metabolites (guidance issued on the website) and the use of the TTC approach for plant and livestock metabolites for the purposes of the Residue Definition for risk assessment. HSE is also working towards increasing its engagement with stakeholders at national and international level on NAMs.
Regulated Products UK
186. Ms Ruth Willis presented an overview of regulated products risk assessment. The role of the FSA is ensuring food you can trust via the organisations core values which are: food is safe; food is what is says it is; and food is healthier and more sustainable.
187. Following EU Exit, the FSA and Food Standards Scotland as the regulators are responsible for approval of all food and feed regulated products in Scotland, England, Wales and Northern Ireland. FSA follow a robust risk analysis process to deliver independent evidence-based advice and recommendations to ministers.
188. As a regulator there is a balancing act between consumer and industry’s desire for new foods and safety and proportionate oversight. Therefore, the FSA ambition is to aim to be a proportionate and agile regulator supporting food innovation while maintaining food safety standards. FSA will look to get the balance right in terms of how much information we need from business applicants to be able to conduct risk assessments and deliver robust and timely regulatory decisions.
189. Regulated products approvals are delivered in partnership with a lot of government departments and scientific advisory committees. The steps in the regulatory framework include:
- Validation - the purpose of which is to ensure the key information for assessment and risk management is provided.
- Assessment - the purpose of which is to review the information provided by the applicant to assess whether the product is safe for the proposed uses and use safe levels.
- Risk management - the purpose of which is to recommend whether to authorise a product and under what conditions.
190. Regulated products as a group cover a broad range of products from food additives to Genetically modified organisms, Feed additives to Food contact materials. They each have their own issues and challenges. For each, there are a set of data requirements.
191. The products are assessed and reviewed by different joint expert advisory groups which advice to the FSA.
192. Key assessment points include:
- What is it? and can they prove it? - composition and specification.
- How is it made?
- What do we know about it from how it has been used before?
- How will it be used? – proposed use and exposure.
- Is it absorbed and where does it go if it does? – ADME.
- Toxicological risks / chemical risks.
- Microbiological risks.
- Nutritional quality (novel foods).
- Allergenicity risks (novel foods).
193. The risk managers then use the risk assessment to identify if there are risks to be managed and make recommendations to ministers on how this can be achieved. This allows consumers to have confidence in the products that reach the market.
International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) Guidelines
194. Dr David Jones presented an overview of the ICH process and its effects on the testing of pharmaceuticals.
195. The need for harmonisation of drug regulations came after preliminary contacts between officials from Europe, Japan and the USA, discussed during the International Conference of Drug Regulatory Authorities.
196. As they all had their own guidelines the discussions revealed the need for harmonization of requirements relating to the new innovative drugs and led to the establishment in 1990 of the International Conference on Harmonisation of Technical Requirements for the Registration of Pharmaceuticals for Human Use (ICH), now called International Council for Harmonisation. ICH meetings are still held twice a year and the UK is a member in its own right.
197. On the ICH website there is a list of all the guidelines.
198. ICH Working Groups are comprised of experts representing ICH Members and Observers. The ICH Harmonisation activities fall into six categories.
199. These activities include:
- The formal ICH procedure.
- Q&A procedure.
- Revision procedure.
- The maintenance procedure.
- Error correction.
- Guideline withdrawal.
200. The selection of a new topic is a formal procedure that can be submitted by any ICH Member or Observer to the Management Committee. The Management Committee (MC) will review any topic proposals received and prioritise proposals to be recommended for endorsement by the ICH Assembly. If the Assembly chooses to endorse the topic that is being proposed for the harmonisation, it will also be asked to endorse the outline for the Concept Paper. In principle, the agreement of all the ICH Members of the Assembly is necessary for initiating any ICH harmonisation activities. However, in exceptional cases Assembly consensus can’t be achieved, and the Assembly will proceed to where a decision to endorse a new topic proposal will be adopted.
201. An informal working group develop a concept paper and business plan which needs to be approved by the Assembly by a formal Expert Working Group (EWG). The ICH Members nominate representatives to the EWGs. Requests received from the WG for additional expertise should be reviewed by the MC on an ad-hoc basis. Most of the EWG work is conducted virtually with face-to-face meetings subject to decision by the MC.
202. When the EWG reaches a consensus on the Technical Document, the ICH Secretariat will organise a Step 1 Experts Sign-Off by the Topic Leaders EWG. The document is then submitted to the Assembly to request endorsement under Step 2a of the ICH process. After, Step 1 has been reached, the WG should provide, to the MC, an estimate of the length of time for the public consultation period in each region. The consensus text of the draft Guideline is endorsed by the Regulatory Members of the ICH Assembly as Step 2b Draft Guideline. At this step, the Step2b draft Guidelines leaves the ICH process and becomes the subject of normal wide ranging regulatory consultation in each of the Members region e.g. for European Commission, Europe it is published as a draft guideline.
203. Each region’s public consultation period may range from 30 days up to several months for more technical Guidelines. After obtaining all regulatory consultation results, the EWG that organised the discussion for consensus building will be resumed. Once the experts from the ICH regulatory Members reach consensus on a revised version of the draft guidelines after consideration of the consultation results. the ICH secretariat will organise a Step 3 sign off by the EWG expert of the ICH Regulatory Members.
204. Step 4 of the ICH process, the Regulatory Members of the Assembly agree on a harmonised guideline in consultation with the MC. This adoption is backed by recommendation by the Management Committee and the consensus of the ICH Assembly Regulatory Members affirming that the Guideline is recommended for the adoption by the Regulatory Members of the ICH regions. Once Step 4 is reached, the harmonised Guideline moves to the final step of the process and is implemented by each of the Regulatory Members in their respective regions.
205. ICH has encouraged greater collaboration among scientific, technical, and medical experts from regulators and companies. It has helped mitigate confusion regarding regulatory processes. ICH has reduced redundancy in requirements at every step of medicine development: in non-clinical studies, clinical trials, submissions, and manufacturing processes. The number of represented countries and regions has considerably expanded from the initial three to now fifty medicine authorities from across the world.
Roundtable Discussions from Session 3
206. Tools: Use of visualisation tools, tables, charts, colour grids e.g. Synthesis and Integration of Epidemiological and Toxicological Evidence (SETE) to summarise data in order to see patterns. However, this still needs to encapsulate considerations for variability.
207. Data: What data are we willing to accept? It was questioned at what point can one accept other bodies’ risk assessments, given that EFSA has significantly more resource than GB and it still struggles with the volume of data that it has for assessment. It was suggested to review other countries (Canada and Australia for example) and what their approach to this issue is. However, issues were highlighted where opinions from other countries are not always accepted by committees due to the lack of original data provided within them, therefore there is reluctance to accept the opinion without such data. It was noted that retrieving this data from industry would be challenging as often this is not easily accessible due to it being deemed confidential. Question of a centralised database of data used in COT papers was raised, possibly one from which graphs and tables may be extracted, although logistically this is an ambitious challenge.
208. Guidance: A guidance for now or a guidance for the future? The key is to find the general principles; however, this may be too open ended for a guidance/guidelines document. Data interpretation is important to include. Development and publication of the general principles also has a drawback in that sub-guidelines/guidance are not aligned, and it is not a one-stop shop. Each will also need to be updated. How prescriptive should guidance be? The participants agreed there would need to be a functional process in order to determine study relevance and that the data should be systematically evaluated for validity- this was indicated as another area in which GB could become a leader. It was suggested that each agency could tell applicants what data they would require; it was noted that if this is not specified there may be an array of different studies received which may not be relevant or fit for purpose. It was further added that this information would also be of value to academic researchers, in helping design and report studies. The weighting of the risk benefit interplay should also be considered as it changes consumers’ behaviour/perception. The guidance should not be too prescriptive and should advocate a case-by-case approach. We still need sufficient information so that industries don't supply negative data which doesn't show a lack of effect because the wrong tests have been done. The UK Committee on Mutagenicity (COM) requires information on the apical endpoints, i.e., chromosome damage, gene mutation, and chromosomal numerical changes. Therefore, depending on what needs to be done, and what substance is being regulated, that’s the type of thing that should be in the guidance. There needs to be certain pieces of evidence, this evidence can come from doing tests. The tests conducted need to be capable of showing a positive effect which may have human relevance.
209. Assessments: Different ways of conducting assessments. It was agreed that a tiered approach is important especially for regulated products, but it was important to consider how new methodologies, such as NAMs, could be integrated alongside this. It was suggested that currently, NAMs were very useful for hazard identification but determining a reference point from data and extrapolating the data to humans was still tricky.
210. Are we only going to accept OECD studies? The participants questioned if only OECD studies should be considered and decided that a variety of studies should be considered, including scientific publications depending on the relevance and level of data, as well as retrospective studies. It was commented that if only OECD studies were to be considered this would rule out NAMs as there are currently almost no OECD guidelines for these studies and implementing these would take a significant amount of time. Do we have to have a mechanistic link for adversity? It was acknowledged that risk assessors and managers and researchers might weight the same information differently based on their interpretation, and it was suggested workshops could be held to bring researchers and regulators together. It was suggested that in future, with much greater use of NAMs, mode of action (MoA)/AOP data will be far more relevant, and this is something we should start looking for more and more. In the past, MoA/AOP has been rather overlooked and now is often a gap in the available data.
211. Systematic reviews were discussed, and that the quality of individual studies included in reviews needs to be assessed. An assumption persists that published papers reach a certain standard, but this is not always the case. These reviews can also be confounded by important variables that are not always considered by the authors, or are not reported in the original studies, such as socioeconomic status. Systematic reviews, moreover, do not include unpublished pieces of work (i.e. non-academic papers) that can contain important pieces of evidence.
212. We will need to work through different scenarios, but also every element as part of different sub-systems, and part of the bigger system. We can apply this kind of thinking to toxicological tests and guidelines i.e. how will the substance interact in different environments? What is the impact in animals, which in turn will affect humans? How will the substance change through this process? How do the different sub-systems in the human body interact? Trying to understand who will be more susceptible and why. This will help us to extrapolate one substance to a class of substances to future materials.
213. Uncertainty: What degree of uncertainty is acceptable? It was suggested that the degree of uncertainty that is acceptable is very much dependent upon the nature of the adversity being assessed.
214. Defining uncertainty is therefore an important practice. Uncertainty is a broad term, that has technical and colloquial meaning across the risk analysis process. The discussions continued as to what might be classified under ‘risk perception’, for instance, how consumers may think home prepared food is ‘safer’ than that purchased outside the home, but the former may contain acrylamide at higher levels through the process of frying/baking etc. Moreover, it was argued that when considering risk, you can’t stop people from doing what they want e.g., smoking, drinking.
215. The definition and degree of uncertainty depends on what we are looking for. If it is a pharmaceutical chemical, then there will need to be a good degree of certainty about whether and how much harm it is likely to cause in patients. However, if it is a chemical in the environment where it is widely used but not within the REACH framework, then a high degree of uncertainty (i.e. conservatism) is acceptable if there is no other information present, and there is no-one willing to pay to generate that information. So, the degree of uncertainty depends on what the chemical is, and what it is being used for.
216. What is the benefit against the risk? For example, consumption of farmed salmon may lead to higher levels of mercury exposure, but that's weighed against the fact that oily fish is good for you. So, we need to look at what is the benefit of the product. For example, an anti-cancer drug could have a higher acceptable level of risk attached to it, because the benefit of taking it will outweigh the risks if the cancer is life-threatening.
217. Uncertainty as a toxicological concept, which can differ from other individuals’ interpretation of ‘uncertainty’ and so messages regarding uncertainty might be misinterpreted. Lay summaries might use different language, but these issues are related to the wider risk analysis process and may be beyond the COT remit. It is prohibitive to address all ‘uncertainties’ in a specific instance. It was commented that if all uncertainties had to be addressed, nothing would be approved for use.
218. Validation: Different organisations are approaching it differently. The people from what is now the DTI (Department of Trade and Industry) have a framework to try to systematise a safety assessment, which includes building works. Should we be looking at everything separately, or whether there is room for looking at everything as a 'one-stop-shop'?
219. We need to think about whether it's a modular system. If it's a modular system, we will need to think about what sub-systems it's made of, the integrity of each sub-system, dependency across the sub-systems, inter-dependence, as well as intended or non-intended impacts of one sub-system. For example, electrical safety can spill into fire safety and structural integrity (because the building can collapse in on itself).
220. Similarly, Good Laboratory Practice (GLP) is not necessarily an index of good science, only that the study was performed to specific standards and can be audited. Combining GLP with a Klimisch score might be a better indicator of study quality.
Session 4: Looking to the future-Moving Forward
Committee on Carcinogenicity (COC) and Committee on Mutagenicity (COM): building bridges
221. Britta Gadeberg and Dr Ovnair Sepai presented on COC and COM guidance. COM and COC are sister committees of COT, jointly sponsored by Department of Health & Social Care (DHSC) and Food Standards Agency (FSA).
222. As for COT, the COC and COM remits cover chemicals in food, consumer products and the environment, but their focus is specifically on carcinogenic and mutagenic potential.
223. COM’s guidance was first written in 1981 with the latest update published in 2021.
224. There are 3 stages of the COM guidance which are:
- Stage 0: Preliminary considerations prior to genotoxicity testing – this covers Structure Activity Relationship (SAR), screening tests and physicochemical properties (of substances and impurities).
- Stage 1: In vitro genotoxicity testing – which includes bacterial gene mutation tests (Ames test) and in vitro micronucleus test for clastogenicity and aneugenicity.
- Stage 2: In vivo genotoxicity tests – which includes a number of tests and considerations to be made with respect to study selection.
225. When updating the guidance, because the science is moving so fast it’s important that you have guidance documents that can be standalone and just update sections as necessary, without having to update the whole document. For example, COM has updated the series of guidance statements with standalone additions of 3D cell models and nanomaterials as well as germ cell mutations. QSAR and quantitative assessment will be looked at in future in order to integrate them better into the assessment process.
226. COC first published its guidance in 1982. In 2012, this was formulated as short specific guidance statements on different topic areas, such as hazard identification and characterisation, cancer risk assessment methods, and guidance on less than lifetime exposure, with an overarching statement of the approach as a whole. COC has a regular review process for each of these and will either issue updates where there have been minor developments or, when required, publish a full revision.
227. The COT and COC Synthesising Epidemiological Evidence Subgroup (SEES) and Synthesis and Integration of Epidemiology and Toxicological Evidence Subgroup (SETE) Reports sit alongside the COC guidance statement series.
228. A number of areas for joint working across the Committees were suggested and include: the use of biomonitoring data in risk assessment, benchmark dose modelling approaches, thresholds, use of NAMs data and consideration of methods, interpretation of margins of exposure, uncertainty factors, exposure assessment, horizon scanning and how to interlink and cross reference between Committee guidance.
Next Generation Risk Assessment (NGRA) and future guidelines
229. Dr Camilla Alexander White presented on policy goals for implementing new Approach Methods (NAMs). The speaker started by noting that we may need evolution in the short term but revolution in the longer term. There will need to be major changes in regulatory thinking.
230. The speaker then stated the reasons why we should use NAMs in chemical policy. Governments and industry must ensure that policies protect people and wildlife from suffering adverse impacts on quality or longevity of life when exposed to chemicals. It should not be the policy goal to align every current toxicological hazard characterisation assay with a NAM, and then validate the NAM against old animal data. To use NAMs there will need to be regulatory reform and new technical guidance by replacing today’s approach with a different approach and the ‘Risk is a function of Exposure and Hazard’ paradigm will stay the same. The nature of the NAM science will influence the design of future regulation and technical guidance; therefore, we should not try to shoehorn NAMs into current regulatory guidance.
231. The way we should see NAMs being used is to help us understand aspects of human exposure and biological mechanisms or mode of action, as part of a classical toxicological risk assessment paradigm. It can also help us generate data related to adverse health effects that have been challenging to model in animals
e.g. developmental neurotoxicity, immunotoxicity, with more human relevant systems.
232. In Next Generation Risk Assessment (NGRA), data can be generated that we can use to make a decision about whether a chemical in a product exposure context is ‘safe’ or presents an ‘acceptable risk’.
233. There is a need for change as the current regulatory paradigm largely protects people, but it is not applying state-of-the-art science, such as animals are not humans and public protection comes from applying crude uncertainty factors in risk assessment to account for that. Toxicological science needs to apply modern biology and chemistry approaches to understand everyday chemical exposure and better understand how chemicals really interact with the human body, diet and medicines etc. Societal views, which are strong in the UK that it is not appropriate to use animals in cosmetics safety assessment, are getting perhaps wider. Innovation can, in principle, be better supported by NGRA with improved understanding of real human exposure and human relevant biology, by data driven exposure assessment which is closer to reality rather than by using worst case assumptions and the NAM data using different uncertainties as well as different uncertainty considerations in risk assessment.
234. The nine principles of NGRA are as follows:
Principle 1: the overall goal in human safety assessment.
Principle 2: the assessment is exposure led.
Principle 3: the assessment is hypothesis-driven.
Principle 4: the assessment is designed to prevent harm.
Principle 5: the assessment follows an appropriate appraisal of existing information.
Principle 6: the assessment uses a tiered and iterative approach.
Principle 7: the assessment uses a robust relevant methods and strategies.
Principle 8: sources of uncertainty should be characterised and documented.
Principle 9: the logic of the approach should be transparent and explicitly documented.
235. The speaker then described where the change is happening. In industry, this is in the cosmetics and agrochemicals sectors. In the USA e.g. US EPA application of NAMs on PFAS assessment and in Europe examples include European Human Biomonitoring Initiative (HBM4EU) and Partnership for the Assessment of Risks from Chemicals (PARC) Project.
236. The key need for a global chemicals policy in the United Nations context is ‘capacity building’ within the new science policy panel for chemicals, waste and pollution prevention over the coming 20 years. Some questions raised were: Does UK want to be sharing our knowledge with developing world partners about the traditional toxicological risk assessment paradigm based on animals? Or the new paradigm based on NAMs?
237. The speaker then proposed a practical revolution for the future. Should we be estimating exposure by measurement and model realistic exposures for the different scenarios for consumer, worker, general population, and environmental species exposure. Should we be investigating how biology is likely to be affected by the level of exposure by using NAMs (and human studies where possible). Should we be doing risk characterisation by new statistical methods, Bayesian approaches, probabilistic approaches; what is the likelihood of adversity at this exposure?
238. Therefore, some next steps should be for industry to share their knowledge with regulators and academia about NAM data generation (including skills development in targeted training courses). More research and skills development on the application of exposure monitoring and modelling is needed. We should consider changing the regulatory paradigm to a ‘NAMs first’ policy direction and provide exposure based or knowledge-based justification why NAMs cannot be used in each case. Then we could draft mock regulatory technical guidance as a research project about how a new exposure-based paradigm would work in practice in NGRA. We should consider how chemicals regulation would need to change with this new science such as GB REACH, Product regulations and COSHH. We should also set a timeline for when regulatory changes need to happen, so we don’t wait for science to be perfect.
239. This would allow the UK to take a global lead and be a beacon based on scientific excellence, creativity, and new thinking.
Evidence synthesis
240. Dr Paul Whaley presented on the evolution of evidence synthesis in environmental health. The speaker started by noting some of the work they have been involved in including for organisations such as the World Health Organisation (burden of disease, GAQGs); US EPA (IRIS phthalates), VKM (in vitro appraisal), EFSA and EBTC (data standards). A lot of the work involves the machinery of producing scientific data which needs to work better: more valid, more shareable, more equitable and ultimately more rewarding.
241. Before 2006 was prehistory, with 2014 kicking off systematic reviews as an approach. In 2019, there was an evolution in evidence synthesising methods relating to evidence mapping techniques and now, we are in an informatics revolution.
242. Lessons learnt include those from narrative reviews conducted by experts, however; what experts think the evidence is saying does not track what the evidence is actually saying. There is insufficient transparency for stakeholders, questions about reproducibility of results and the Bisphenol A (BPA) controversy is a good example of this.
243. So, the first need was for reproducible methods for assessing exposure-outcome relationships which drove the uptake of systematic reviews. Lots of different definitions of systematic reviews exist, however the preferred one is applying scientific method to using existing evidence to answer research questions instead of conducting a new study. The origins of the approach are in medicine and social sciences, first being mooted for toxicology around 2006. In 2010, EFSA published a navigation guide Application of systematic review methodology to food and feed safety assessments to support decision making. In 2014, the National Toxicology Program published Handbook for Conducting a Literature-Based Health Assessment Using OHAT Approach for Systematic Review and Evidence Integration. It was then formalised in the Environmental Health Protection Agency (EPA) ORD Staff Handbook for Developing IRIS Assessments in 2021.
244. Systematic reviews are good for specific questions, but not good for broad questions. There is a lot of work, so prioritisation is key; however, it is difficult to know what to prioritise transparently. The effort you put in has regulatory consequence and it should and does require transparency.
245. Therefore, the second need is for transparent, objective methods for surveying domains of evidence. There needs to be specific questions about exposure-outcome associations.
246. It requires systematic mapping methods, which are applying systematic review techniques to scoping reviews. It is a geography of the evidence base so you can prioritise clusters and gaps: this is a concept that came from social science and environmental research around 2015. Then it was adapted to environmental health with NTP around 2019. It was then integrated into US EPA IRIS assessment processes in 2021. It is important to note from this process that those products needed to be interoperable and made tangible the idea of a single database to mapping an entire domain.
247. The third need is that research data should be Findable, Accessible, Interoperable, and Reusable (FAIR). The informatics revolution is currently happening, which has open standards for storing and retrieving research data. Lessons learnt from this is that there is too much information, and processing needs to be automated by making research data computable. This can be done by using Natural Language Processing (NLP) techniques to abstract and classify data (applying open data standards retrospectively); and move away from the paper-based paradigm of research (integrate open data standards into primary research processes).
248. The speaker then discussed what it meant for committees like the COT. There are big changes happening fast. There will be profound implications for governance and decision-making so committees will need to be aware of these but perhaps need not address them themselves. There will be a lot of automated evidence monitoring systems. There will be a need for assessment triggers, rationalised structures for responding and there will be a need to systematise the prioritisation of questions and resources.
Roundtable Discussions from Session 4
249. AI and ChatGPT: Artificial Intelligence and machine learning as an aid for this kind of data interrogation and manipulation were briefly raised. However, which data would train such algorithms was the main challenge identified. Members suggested ChatGPT would be a great way to search a high volume of information but that it must be paired with human enquiry as it cannot be fully trusted if it hallucinates. Regarding WoE vs quantity vs quality, members agreed that quality is preferred over quantity. Members stated that literature should be analysed for its relevance and value in terms of scientific content. One member questioned who determines what quality is; it was explained that there are numerous scientific means of analysis which can be undertaken that deem a piece of research to be of high quality. The final discussion entailed members agreeing that methodologies would need agreeing for risk benefit analysis, and stated this has proven to be difficult due to not being able to compare risk and benefit using a comparable term or approach.
250. Weight of Evidence vs quantity vs quality: Quality might be compromised if the studies didn't use the newest approaches. Quantity doesn't really count, unless there are some good studies in the overall database. We need to qualify each and every study, and then group those studies according to their relevance and value of their scientific content. So overall, it’s best to have a WoE approach that is based on the quality of each study and also the quantity of good studies. There are quality criteria we can apply (such as study design, and blinding) in order to distinguish a good study from a bad study. Then you have a group of experts, and based on their collective wisdom they arrive at a qualitative/ quantitative answer based on all of the studies they have. Using a WoE approach can mean collecting a lot of very similar papers. However, it is also possible to get different strands of evidence to establish a causal relationship.
251. When you use WoE without having defined it, the WoE approach becomes complicated. So, you need to identify how you will assess the WoE first. For example, having a protocol which says how you will collect the data, then how you will appraise and evaluate the studies, and then how will this be integrated? The advantage of a systematic approach is that you can see which studies have been assessed, which studies have been discarded and why. It’s still possible to amend the protocol and explain why it was amended in a transparent way. Objective suitability criteria are needed in order to evaluate each study, e.g. how reliable is it? And then how does it play into the evidence-base using those quality criteria?
252. Comparative risks? Risk benefit?: In terms of providing dietary advice, the regulator will need to look at the full picture. The methodologies would look at both risks and benefits in an objective manner, such as for e-cigarettes, plant-based drinks. EFSA is currently working on an updated guidance for risk-benefit assessment. However, developing this new guidance is proving to be difficult, because it is difficult to quantify risks and benefits. Current approaches include life years disease-adjusted for 'lost years', but this approach is not fully applicable here. For example, what is the overall benefit of eating oily fish, and how much of the fish do you eat? Ultimately, we need tools to be able to assess the risk/benefit on various levels, from the 1:1 comparisons, up to the whole-food assessment.
253. Food is the most difficult problem to solve, as it depends on supply, and the different nutrients that people need. It makes the assumption that food is readily available to everybody, which is not necessarily the case. The way different people proportion their diet is not necessarily the same, not just through personal preference e.g. vegetarianism, but it could be due to a shortage of food. If you have, say a vegan diet, then you change the benefits and the risks. Personal choices are also affected by affordability.
254. If a chemical needs to be added to food because it brings a certain benefit, then it becomes a regulated product and it is relatively easy to do a risk/benefit analysis. But what should be done if instead you have contaminants? Do you accept the health risk from the contaminants because you know there is also a benefit from the food in which it is present?
255. It was also noted that comparative risks may be a useful concept, if for example there's a safer additive to food. However, the example of e-cigarettes is less clear. Products such as e-cigarettes contain nicotine. E-cigarettes are being marketed as a safer alternative to smoking tobacco, but the flavours being used appeal to children/young people and their use increases the probability that children will become addicted to nicotine.
256. Training and Skills: A large issue identified by the group was the current skills shortage across both GB and Europe, where there are a very limited number of people trained and skilled as environmental health and toxicology scientists. A key aspect highlighted for moving forward with guidance and the COT was that more people will need to be educated and upskilled in order to populate secretariats and committees with the relevantly skilled people, however it was acknowledged that this comes down to funding available on a wider scale.
Conclusions and recommendations
257. The workshop served as the kick-off to update COT’s guidance on the assessment of chemical toxicity and its supporting principles, starting with existing frameworks and guidance, but with the aim of ensuring that the guidance reflects the state-of-the-science.
258. The workshop aimed to identify those key areas where the guidance most needs to evolve, which included food safety goals, current guidance on risk assessment and what can be learned from it, integration of new approach methodologies, exploring hazard vs risk and weight of evidence.
259. The workshop identified and reviewed a number of important topics that would need to be covered in the guidance, including models and tools, assessment methods, data sources and quality, uncertainty, new approach methodologies (NAMs), training and skills, validation, regulatory interface, and the role of scientific advisory committees (Figure 3).
This figure contains the workshop report logo with text dotted around it titling themes from the workshop. The logo is in colour and the text is black.
Figure 3. Themes from the workshop.
260. The workshop’s recommendations, priorities and proposed improvements to the COT guidance are illustrated in Table 1.
Table 1. Recommendations, priorities, and proposed improvements to the COT guidance from the “Evolving our assessment and future guiding principles” workshop.
|
Recommendations |
Priorities |
Improvement |
|
Transparency |
Resourcing and capturing institutional wisdom |
Clear distinction between who is responsible for what |
|
Build relationships with other agencies and harmonization internationally |
Weight of Evidence |
Strengthen links between Scientific Advisory Committees across government agencies
|
|
Explore uncertainty and validation |
Tiered Testing |
Integrated (systematic) reviews in risk assessment |
|
Explore risk benefit and comparative risk |
Enhancing public health |
Codify what we have |
Post Workshop Discussions and steps going forward
261. The Committee reviewed the workshop report and Table 1 and identified the must, should and could most important areas for the future.
Must: Identify and adapt existing frameworks, guidance, and assessments to enhance public safety.
Should: Integrate new approach methodologies by using the best available science.
Could: Explore risk benefit and comparative risk.
262. The committee concluded that the most important aim going forward was to have applicable guidance for the Committee that ensures consumer safety, based on fundamental risk assessment principles, using the best science available.
References
Barlow, S.M., Greig, J.B., Bridges, J.W., Carere, A., Carpy, A.J.M., Galli, C.L., Kleiner, J., Knudsen, I., Koeter, H.B.W.M., Levy, L.S. and Madsen, C., 2002. Hazard identification by methods of animal-based toxicology. Food and Chemical Toxicology, 40(2-3), pp.145-191.
Cattaneo, I., Kalian, A.D., Di Nicola, M.R., Dujardin, B., Levorato, S., Mohimont, L., Nathanail, A.V., Carnessechi, E., Astuto, M.C., Tarazona, J.V. and Kass, G.E., 2023. Risk Assessment of Combined Exposure to Multiple Chemicals at the European Food Safety Authority: Principles, Guidance Documents, Applications and Future Challenges. Toxins, 15(1), p.40.
COT/ FSA Paving the way for a UK Roadmap: Development, Validation and Regulatory Acceptance of New Approach Methodologies (NAMs) in Chemical Risk Assessment (Draft Version 3) Available at: UK New Approach Methodologies Roadmap (2023) Draft Version 3 | Committee on Toxicity (food.gov.uk)
European Food Safety Authority Guidance on the use of the benchmark dose approach in risk assessment. Available at: Guidance on the use of the benchmark dose approach in risk assessment | EFSA (europa.eu)
European Food Safety Authority Guidance and other assessment methodology.
Available at: Guidance and other assessment methodology documents | EFSA (europa.eu)
European Food Safety Authority Guidance on the use of the weight of evidence approach in scientific assessments. Available at: Guidance on the use of the weight of evidence approach in scientific assessments | EFSA (europa.eu)
European Food Safety Authority Guidance on harmonised methodologies for human health, animal health and ecological risk assessment of combined exposure to multiple chemicals. Available at: Guidance Document on Scientific criteria for grouping chemicals into assessment groups for human risk assessment of combined exposure to multiple chemicals | EFSA (europa.eu)
European Food Safety Authority Guidance Document on Scientific criteria for grouping chemicals into assessment groups for human risk assessment of combined exposure to multiple chemicals. Available at: Guidance Document on Scientific criteria for grouping chemicals into assessment groups for human risk assessment of combined exposure to multiple chemicals - - 2021 - EFSA Journal - Wiley Online Library
European Food Safety Authority Risk to human health related to the presence of perfluoroalkyl substances in food. Available at: Risk to human health related to the presence of perfluoroalkyl substances in food | EFSA (europa.eu)
European Food Safety Authority Application of systematic review methodology to food and feed safety assessments to support decision making. Available at: Application of systematic review methodology to food and feed safety assessments to support decision making | EFSA (europa.eu)
Health and Safety Executive Meeting the requirements for toxicological information in applications for authorisation of Plant Protection Products under Regulations (EC) No 1107/2009 and (EC) No 284/2013 (2022). Available at: Toxicology - Plant Protection Products (hse.gov.uk)
IPCS Harmonization Project. Available at: International Programme On Chemical Safety Harmonization Project Strategic Plan (who.int)
Kotthoff, M., Fliedner, A., Rüdel, H., Göckener, B., Bücking, M., Biegel-Engler, A. and Koschorreck, J., 2020. Per-and polyfluoroalkyl substances in the German environment–levels and patterns in different matrices. Science of The Total Environment, 740, p.140116.
National Toxicology Program Handbook for Conducting a Literature-Based Health Assessment Using OHAT Approach for Systematic Review and Evidence Integration.
Available at: Handbook for Conducting a Literature-Based Health Assessment Using OHAT Approach for Systematic Review and Evidence Integration (nih.gov)
Linkov, I., Loney, D., Cormier, S., Satterstrom, F.K. and Bridges, T., 2009. Weight-of-evidence evaluation in environmental assessment: review of qualitative and quantitative approaches. Science of the Total Environment, 407(19), pp.5199-5205.
Renwick, A.G., Barlow, S.M., Hertz-Picciotto, I., Boobis, A.R., Dybing, E., Edler, L., Eisenbrand, G., Greig, J.B., Kleiner, J., Lambe, J. and Müller, D.J.G., 2003. Risk characterisation of chemicals in food and diet. Food and Chemical Toxicology, 41(9), pp.1211-1271.
World Health Organisation Principles and methods for the risk assessment of chemicals in food (2009). Available at: Principles and methods for the risk assessment of chemicals in food (who.int)
Abbreviations
|
Adverse Outcome Pathways |
AOPs |
|
acute toxicity estimate |
ATE |
|
Aryl Hydrocarbon Receptor |
ARH |
|
Benchmark dose modelling |
BMD |
|
Benchmark Response |
BMR |
|
The Committee on Toxicity of Chemicals in Food, Consumer Products and the Environment |
COT |
|
Panel on Contaminants in the Food Chain |
CONTAM |
|
Department of Health and Social Care |
DHSC |
|
Department for Environment, Food & Rural Affairs |
DEFRA |
|
European Chemicals Agency |
ECHA |
|
Environmental Health Criteria monographs |
EHCs |
|
European Food Safety Authority |
EFSA |
|
Food Standards Agency |
FSA |
|
General Data Protection Regulation |
GDPR |
|
health based guidance values |
HBGVs |
|
Health and Safety Executive |
HSE |
|
International Agency for Research on Cancer |
IARC |
|
International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use |
ICH |
|
International Programme on Chemical Safety |
IPCS |
|
margin of exposure |
MOE |
|
Molecular Initiating Event |
MIE |
|
Mode of Action |
MoA |
|
New Approach Methodologies |
NAMs |
|
National Toxicology Program |
NTP |
|
National Institute of Environmental Health Sciences |
NIEHS |
|
Northern Ireland Protocol |
NIP |
|
Occupational Safety and Health Administration |
OSHA |
|
Organization for Economic Cooperation and Development |
OECD |
|
persistent, bioaccumulative and toxic |
PBT |
|
Perfluoroalkyl and Polyfluoroalkyl Substances |
PFAS |
|
Perfluorononanoic acid |
PFNA |
|
Perfluorohexanesulphonic acid |
PFHxS |
|
Perfluorooctane sulfonate |
PFOS |
|
Perfluorooctanoic acid |
PFOA |
|
points of departure |
POD |
|
Registration, Evaluation, Authorisation and Restriction of Chemicals |
REACH |
|
Reference Point |
RP |
|
Scientific Advisory Committee |
SAC |
|
Tolerable Weekly Intake |
TWI |
|
very Persistent and very Bio-accumulative |
vPvB |
|
Weight of Evidence |
WoE |
|
World Health Organisation |
WHO |
Organizing Committee
Dr Olivia Osborne
Ms Cath Mulholland
Miss Sophy Wells
COT Members
Professor Alan Boobis (Chair)
Dr Phil Botham
Ms Jane Case
Dr Stella Cochrane
Dr James Coulson
Dr Silvia Gratz
Professor Thorhallur I. Halldorsson
Professor Gary Hutchison
Dr Sarah Judge
Professor Gunter Kuhnle
Dr David Lovell
Professor Shirley Price
Dr Mac Provan
Ms Juliet Rix
Dr Michael Routledge
Dr Cheryl Scudamore
Dr Natalie Thatcher
Professor Mireille Toledano
Dr Simon Wilkinson
Professor Philippe Wilson
Professor Matthew Wright
Professor Maged Younes
COT Secretariat
Ms Catherine Mulholland
Ms Britta Gadeberg
Dr David Gott
Dr Alexander Cooper
Dr Barbara Doerr
Ms Jocelyn Frimpong Manso
Ms Cleanncy Hoppie
Mr Barry Maycock
Dr Olivia Osborne
Ms Claire Potter
Ms Sabrina Thomas
Ms Chara Tsoulli
Ms Frederique Uy
Miss Sophy Wells
Dr Gaetana Spedalieri
Dr Michael Dickinson
Mr Thomas Hornsby
Ms Gail Drummond
Dr Emily Hudson
Dr David Kovacic
Ms Kaitlyn Jukes
Dr Aaron Bradshaw
Dr Lorcan Browne
Ms Natasha Adams
Ms Abigail Smith
Dr Katie Schulz
Ms Katie Wetherall